Last Updated on October 29, 2023
Dysplasia epiphysealis punctata is a rare condition characterized by the presence of stippled epiphyses at birth in association with other connective tissue disturbances.
It is also known under a variety of names such as chondrodystrophia foetalis calcificans, stippled epiphyses, or congenital multiple epiphyseal dysplasia.
It is also called Conradi Disease.
Two main forms are recognized
- The classic, but rarer, severely rhizomelic type due to an autosomal recessive gene
- Conradi-Hunermann variety due to an autosomal dominant gene or a mutant gene
Clinical Features of Dysplasia Epiphysealis Punctata
In the milder forms, dwarfing at birth is not very marked and there may only be non-specific features, notably a mongoloid facial appearance with snub nose with flattening, upward slanting of the eyes and short neck and trunk becoming more obvious as the child grows.
Skeletal deformities are mild without significant contracture at birth and later, minor wrist or metacarpal changes or pes planovalgus. Cataract, skin changes and other general abnormalities are less common in the milder form and deformity may be confined to a single limb or part of it.
Survival into adult life is likely.
In severe cases, there is marked congenital dwarfing, with trunk and limb shortening particularly affecting the proximal segments of the limbs. There are flexion contractures of the hip, knee and elbow joints, a small chest and a saddle-nose deformity.
The limb shortening is often asymmetrical, becoming more obvious with growth and the spine may develop scoliosis or kyphoscoliosis.
Ectodermal changes with alopecia and coarse hair, cataract, skin manifestations such as ichthyosis, hyperkeratosis, erythrodermia or desquamation are common and there may be diminished intelligence.
Congenital cardiac abnormalities are sometimes present, leading to early death. Apart from joint deformities associated with periarticular fibrosis and calcification, other cartilaginous structures such as the nasal septum, trachea or larynx are similarly affected.
Radiological Features
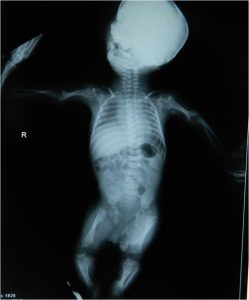
Presence of stippled epiphyses at birth is pathognomic. The size and distribution of the calcification vary from pin-head to lead-shot and from a few specks to extensive areas, usually confined to the region of the cartilaginous epiphyses but sometimes extending to surrounding soft tissues.
The most frequently affected epiphyses are the upper and lower femoral, upper tibial, upper humeral, carpal and tarsal, vertebral and pelvis.
The extent of stippling at birth is not related to the degree of subsequent bone shortening. The abnormal calcification is also seen in the larynx, trachea or nasal cartilages. During the first three years, the calcifications are absorbed.
In mild cases, the epiphyses become normal or almost normal but there is a shortening of the femur and the humerus. The carpal and tarsal bones are often the last to show delayed ossification and stippling. The spine shows alternation in the form of the vertebral bodies, often with blunting of the anterior part of the bodies.
In severe cases, the femora and humeri are short and curved at birth, often asymmetrically. There may be dislocation of the hip and other joint deformities such as coxa vara dislocated patella or scoliosis which develop with growth. In early life, the vertebral bodies may show a coronal cleft.
When the child is first examined radiologically in mid or late childhood, it may be difficult to differentiate this condition from multiple epiphyseal dysplasia or spondylo-epiphysial dysplasia.
Pathology
The cartilaginous epiphyses show patchy mucoid degeneration and cystic spaces with irregular vascularization. There is scattered calcification with poor alignment of cartilage columns and absence of a zone of provisional calcification. Juxta-articular tissues show ectopic calcification.
Progression
In severe cases, more than half the children die within the first year of life from secondary infections. Contractures present at birth tend to improve and curved bones may correct spontaneously, though deformity may recur later.
Treatment of Dysplasia Epiphysealis Punctata
In infancy, passive joint movements, or splints for hip dislocation or foot deformities suffice until the natural progress of the disease is apparent.
If the child survives but is severely affected, surgery may be needed for correction of deformity by soft-tissue release or, in later childhood, by osteotomy, but decisions to operate should be delayed until it is obviously necessary. The skin is often of poor quality and liable to infection.
The spine that develops scoliosis may be especially difficult to manage. An absent vertebral body resulting in severe and progressive kyphoscoliosis may require spine fusion in childhood, as may scoliosis due to unsegmented bars and there is a high incidence of pseudarthrosis.
If there are no bony vertebral abnormalities, a Milwaukee brace can be used to delay or diminish progressive curvature.
Correction of unequal limb length, which is usually due to femoral shortening, by lengthening procedures, should be considered with caution because of the liability to pseudoarthrosis.
In the severest cases, with cerebral defects, surgery must usually be limited to the management of hip adduction deformities by adductor release to allow easier nursing and to try to prevent dislocation.