Last Updated on September 13, 2020
First described as a syndrome by Apert in 1906 Apert syndrome primarily affects the head, hands, and feet and is characterized by fusion or synostosis [fusion] of the cranial sutures and varying degrees of complex syndactyly of the hands and feet.
It is also called acrocephalosyndactyly, type 1 or ACS1.
Apert syndrome is very rare, probably occurring in one in 200,000 births.
Etiology is not known. It is, a genetic disturbance with a strong dominant inheritance but sporadic cases due to mutation do occur.
Cause and Pathophysiology of Apert Syndrome
What is the Cause of Apert Syndrome?
Apert syndrome is caused by mutations in the gene encoding the fibroblast growth factor receptor 2 (FGFR2) protein on chromosome 10q26. These mutations occur randomly [almost exclusively in males]
FGFR2 protein is expressed in
- Cartilage
- Osteoprogenitor cells [type of osteogenic stem cells]
- Connective tissue of limbs
- Skin
- Brain
It has a role in signal transmission from the extracellular environment to intracellular pathways that are important regulators of cell reproduction, differentiation, and apoptosis [programmed cell death].
Serine252Trp and Pro253Arg are major missense mutations responsible for Apert syndrome cases.
Mutations of the human FGFRs have also been identified as the cause of other craniosynostosis syndromes like Crouzon syndrome and others, and skeletal dysplasias such as achondroplasia.
Normal Skull in Children
Apert syndrome results in the closure of coronal suture in infancy. Details of various sutures and their normal behavior are discussed first.
Skull is actually formed by the connection of two frontal, two parietal, and one occipital bone. These bones are plate-like structures and are held together by fibrous lines like joints called sutures.
The purpose of the sutures is to allow bone movements during the birth process and after that allow the even enlargement of skull as the brain grows.

The following sutures are present
- Metopic suture extends from the top of the head down the middle of the forehead, toward the nose at the meeting of two frontal bones.
- Coronal suture extends from ear to ear and connects frontal bones with parietal bones.
- Sagittal suture extends from the middle of the top of the head to the back and connects two parietal bones.
- Lambdoid suture extends across the back of the head and connects parietal bones with the occipital bone.
Sutures fuse by age 24 years. Early fusion of any of the sutures stops the expansion of the skull in that region and compensatory expansion occurs in another area. This results in a head that is not shaped normally and is called (craniosynostosis).
Fontanelles are the space between the bones of an infant’s skull where the sutures intersect) that are covered by tough membranes for protection.
The anterior fontanelle or soft spot is the junction where the 2 frontal and 2 parietal bones meet. It closes at 18-24 months of age.
The posterior fontanelle is the junction of the 2 parietal bones and the occipital bone. It closes earlier than anterior fontanelle.
What Happens in the Apert Syndrome
The coronal suture region is closed during early infancy.
This results in widening of anterior and posterior fontanelles, metopic suture and sagittal suture area in an attempt to accommodate the growing brain There is also the premature fusion of the sutures at the base of the skull which leads to
- Maxillary hypoplasia
- Shallow orbits
- Short nasal dorsum
Occasionally, shortening can lead to upper airway obstruction.
Various Changes Observed in Body are [Not exhaustive list]
- Skull and face
- Skull deformities such as acrocephaly [high peaked skull], high prominent forehead, etc
- Cloverleaf skull [rare]
- Flattened, asymmetrical face
- Low-set ears
- Conductive hearing loss When stapes is fixed]
- Eyes
- Wide separation [hypertelorism], shallow orbits
- Exophthalmos
- Keratoconus
- Optic atrophy
- Congenital glaucoma
- Oral
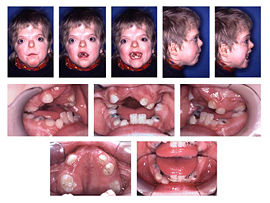
- High arched palate, a bifid uvula, and a cleft palate.
- Crowded upper teeth, misshapen teeth, and other dental issues
- Malocclusion
- Delayed dentition
- Extremities and digits – [Upper limbs are more affected than lower limbs]
-
- Syndactyly [Mitten hands and socks feet]
- Synonychia [contiguous nailbeds]
- Short broad thumbs deviated radially
- Stiffness of shoulder and elbow
- Shortened humerus bones bilaterally
- Central nervous system
- Cognition ranges from normal Intelligence to mental retardation
- CNS malformations
- Increased intracranial pressure
- Other defects
- Congenital cervical spinal fusion
- Tracheal cartilage anomalies
- Skin hypopigmentation, dimples
- Plantar hyperkeratosis in the plantar surface
- Excessive skin wrinkling of forehead
- Visceral abnormalities – cardiovascular, gastrointestinal, genitourinary, and respiratory anomalies
hand Anomalies are discussed in greater detail further
Types of Hand Anomalies
Hand anomalies in Apert syndrome have the following common features
- Abnormally shaped proximal phalanx [delta phalanx] leading to radial deviation of thumb
- Complex syndactyly of the index, long, and ring fingers
- Symphalangism of the central segments of the index, long, ring, and small fingers
- Simple syndactyly of the web space between the ring and small fingers.
The extent of syndactyly between thumb and index finger is the basis for the classification of Apert syndactyly into 3 types
- Type I [Spade Hands]
- Most common and the least severe
- Index, middle, and ring fingers fused
- Thumb free
- Flat palm
- Type II [Mitten or Spoon Hands]
- Thumb also fused along with the fusions mentioned above
- Concave palm
- Type III [Rosebud or Hoof hands]
- Most severe form but least common
- All digits fused with one nail
- The thumb of the hand is turned inward, and indistinguishable
Clinical Features of Apert Syndrome
The head is peaked and vertically elongated in its anteroposterior diameter, with the planes of the face and the back of the skull being parallel.
The enlarging brain causes increased intracranial pressure.
Eyes are protuberant eyes and wide-spaced. There is a divergence of the transverse axis of eyes. Strabismus and progressive impairment of vision are common.
Often the posterior palate is high-arched, and there are fusion defects of the maxilla and mandible.
Convolutional atrophy of the brain and mental retardation are common.
The hand deformities are of variable severity, as described above.
Often both bones of the forearm are shortened, the elbows are stiff, and shoulder abduction is limited.
Diagnostic Work-Up of Apert Syndrome
Prenatal Diagnosis
When parents are affected, there is a minor risk of the disease in their siblings.
Prenatal diagnosis can be made on the basis of abnormal skull or hand findings of the fetus on ultrasound especially by the use of 3D ultrasonography in the second or third trimester.
Prenatal molecular diagnosis can be done by direct DNA testing of fetal samples in amniocentesis or chronic villus sampling to look for the FGFR2 gene.
Genetic testing for FGFR2 mutations has highly sensitive and specific.
X-rays
Skull
- To evaluate skull deformities
- Common findings are
- Sclerosis of the suture line
- Beaking along the suture line
- Indistinct suture line
- Skull shape abnormalities
- Shallow orbits
- Hypoplastic maxillae
Spine
- Cervical fusions mostly C3-4 and C5-6
- Small-sized vertebral body
- Reduced intervertebral disc space
Extremities
- Multiple epiphyseal dysplasias
- Short humeri
- Glenoid dysplasia [abnrormally formed]
- Hands/Feet
- For evaluating the completeness of syndactyly
- Multiple progressive synostoses
- Symphalangism of interphalangeal joints .
- Shortened and radially deviated distal phalanges
- Delta-shaped deformity of distal phalanx of thumb
CT/MRI
CT is able to tell the precise anatomy of the structures involved and MRI is good at delineating soft tissue structures.
Differential Diagnosis
- Crouzon syndrome
- Pfeiffer syndrome
- Saethre-Chotzen syndrome
- Carpenter syndrome
- Jackson-Weiss syndrome
Treatment of Apert Syndrome
Apert syndrome involves many systems and therefore the treatment requires multidisciplinary work.
Medical care of Apert syndrome includes
- Management of eye ailments
- Lubricants
- Protection of the cornea
- Airway care
- Psychological and emotional care
Surgical treatment of Apert syndrome includes the following
- Eyes
- Orbital surgery proptosis, reduction of hypertelorism
- Surgery for corneal protection
- Sleep apnea: Tracheostomy is indicated in severely affected children.
- Surgery for chronic middle ear
- Tracheostomy for severe sleep apnea
- Cranial surgery to remove synostotic sutures and reshape the skull
- Allows more normal cranial development
- Relieves increased intracranial pressure
- Nasal reconstruction surgery
- Facial surgery for normalization of appearance
- Mandibular osteotomies to improve dental structures
- Surgical separation of digits for syndactyly
- Shunting for intracranial pressure rise
Following are the preferred times for various surgeries
- Removal of synostotic sutures – Early infancy, as early as 3 months
- Fronto-orbital advancement – 6-12 months old.
- Midface surgery – around adolescence
- Syndactyly
- First stage – 9-12 months
- Second stage Around 10 years
References
- Cohen MM, Kreiborg S. Skeletal abnormalities in Apert syndrome. Am J Med Genet 1993 (47): 624-32.
- Liu, C, Cui, Y, Luan, J, Zhou, X. The molecular and cellular basis of Apert syndrome”. Intractable Rare Dis Res. 2013 (2): 115-22
- Conrady CD, Patel BC, Sharma S. Apert Syndrome. StatPearls. 2020 Jan.
- Fearon JA, Podner C. Apert syndrome: evaluation of a treatment algorithm. Plast Reconstr Surg. 2013 Jan. 131(1):132-42.
- Barnett S, Moloney C, Bingham R. Perioperative complications in children with Apert syndrome: a review of 509 anesthetics. Paediatr Anaesth. 2011 Jan. 21(1):72-7.