Last Updated on October 29, 2023
Enchondromatosis: insights on the different subtypes
Enchondromatosis is the presence of multiple enchondromas.
Enchondromas are benign, and often asymptomatic lesions of hyaline cartilage in the metaphyses and diaphyses of the short and long tubular bones of the limbs. They are especially common in hands and feet.
They are often found as incidental lesions, usually solitary on the radiological examination for some other reason.
When there are multiple enchondromas, the condition is called enchondromatosis.
Classification of Enchondromatosis
Following are the main types of enchondromatosis depending on the pattern of distribution, genetic transmission, and associations.
No spinal involvement
Non-hereditary
- Ollier disease
- Mafucci syndrome
Autosomal dominant
- Metachondromatosis
- Enchondromas with osteochondroma-like lesions
- Genochondromatosis
- Symmetric with normal stature
- Type 1 clavicular thickening
- Type 2 normal clavicle
Spinal Involvement
Autosomal Recessive
- Spondyloenchondrodysplasia
Unknown mode [recessive or dominant]
- Cheirospondylochondromatosis
- Dysspondyloenchondromatosis
How Does Enchondromatosis occur?
The mechanism of development of enchondroma is largely elusive.
Enchondromas arise in the metaphysis in close proximity to the growth plate and are thought to be the result of terminal differentiation of growth plate chondrocytes.
About 10% of patients with enchondromatosis have a mutation in the receptor, PTH1R, which acts as a receptor for parathyroid hormone and for the parathyroid hormone-related peptide.
Some studies have reported that genes affecting heparan sulfate pathways are also affected.
Ollier disease
Ollier disease is the most common type of enchondromatosis. It was first detailed in 1889.
It is non-hereditary and is characterized by the presence of multiple enchondromas with asymmetric distribution.
Ollier disease is encountered early in childhood and affects both genders equally. Few cases of familial occurrence have been reported.
Often the lesions involve one side of the skeleton. It is most common in short tubular bones of hands and feet followed by long bones like humerus and femur. It is known to affect axial skeleton less commonly but the skull and vertebral bodies are very rarely involved.
Malignant transformation is estimated to occur in 5–50% of the patients. It often occurs in long tubular and flat bones while this is far less common in the small bones of hands and feet.
Intracranial tumors of glial origin are other associations with Ollier disease.
The cause is not known and a somatic mosaic mutation is thought to be responsible
Thus, PTH1R mutations may contribute to the disease in a small subset of Ollier patients but are probably not causative for the disease.
[Read more about Ollier’s disease]
Maffucci’s syndrome
Maffucci’s syndrome is enchondromatosis identical to that of Ollier’s disease coexists with extraskeletal, predominantly soft tissue angiomatosis typically involving viscera, such as the lungs and liver.
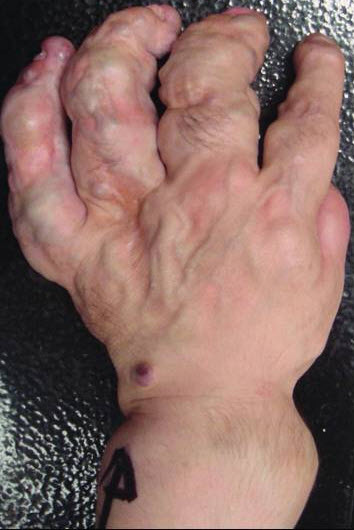
Maffucci syndrome is also called
- Dyschondrodysplasia with hemangiomas
- Enchondromatosis with multiple cavernous hemangiomas
- Kast syndrome
- Hemangiomatosis chondrodys-trophica
It was first described in 1881.
It is rarer than Ollier disease.
Vascular lesions in Maffucci’s syndrome most typically represent cavernous hemangiomas with frequent thrombosis and calcified thrombi, which are often seen on plain radiographs as phleboliths. Hemangiomas in Maffucci’s syndrome tend to be more pronounced in the areas of skeletal involvement by enchondromatosis but are not restricted to these regions.
Often the skeleton lesions manifest earlier and the disease initially may be labeled as Ollier’s disease. Later, vascular lesions may develop.
Apart from hemangiomas, aneurysms, lymphedema, and lymphangiomas may occur. Nodular goiter and adrenal insufficiency malignancy in Maffucci’s syndrome can be associated.
The risk of malignant transformation is even higher than that in Ollier’s disease. Intracranial tumors may be associated
Metachondromatosis
In metachondromatosis, there are multiple enchondromas and multiple osteochondroma-like lesions.
Interestingly, the enchondromas mainly involve the iliac crests and metaphyses of the long bones of the lower extremities whereas the osteochondroma-like lesions are mainly located in hands and feet.
The syndrome can present in early childhood.
The usual osteochondromas point away from joint but osteochondroma-like lesions in the metachondromatosis point towards the joint.
These lesions are not associated with bony deformities.
The disease is extremely rare and transmitted in autosomal dominant fashion.
Genochondromatosis
- Extremely rare
- Autosomal dominant
- Manifests in childhood
- Stature is normal
- Symmetrically distributed enchondromas
- Metaphysis is a characteristic location
- No spinal involvement
- Two subtypes
- Type I involves clavicle
- Type II involves short tubular bones of the hand, wrist, and feet but clavicle spared
- No bony deformities
- Tend to regress in adulthood
- No malignant transformation reported
Spondyloenchondrodysplasia
- An autosomal recessive inherited disorder
- Higher prevalence in Israel
- Usually manifests from birth to infancy
- Vertebral dysplasia combined with enchondromas like lesions in the pelvis or long tubular bones.
- Features
- Platyspondyly – Flat, often rectangular vertebral bodies on X-ray
- Irregular areas of increased and decreased mineralization
- Short broad ilia.
- Short stature (short limbs)
- Increased lumbar lordosis
- Kyphoscoliosis
- Genu valga or vara
- Facial anomalies
- Barrel chest
- Clumsy movements.
- Two types
- Type I – classic
- Type II – Also affects the central nervous system
- Spasticity
- Developmental delay
- Late-onset cerebral calcifications
The spine is less severely affected as compared with dysspondyloenchondromatosis and cheirospondyloenchondromatosis.
There is also an autosomal dominant inheritance pattern that has been reported for this disease.
Dysspondyloenchondromatosis
It is a non-hereditary disorder characterized by spondyloenchondromatosis and spinal malformation.
Presence of irregular vertebral anomalies differentiates dysspondyloenchondromatosis from other enchondromatosis with spinal involvement. The disorder manifests itself at birth.
Features are
- Vertebral abnormalities
- Severe segmentation abnormalities
- Secondary deformities of the vertebral column can be seen
- Neonatal dwarfism
- Unequal limb length
- Asymmetric limb shortening
- Flat midface with a frontal prominence
- Progressive kyphoscoliosis can be found
- Multiple enchondromas
- Long tubular and flat bones
- Bones of hands and feet not affected or mildly affected.
Malignant transformation has not been reported.
Cheirospondyloenchondromatosis
It has symmetrically distributed multiple enchondromas with marked involvement of metacarpals and phalanges.
Short hands and feet are seen with mild platyspondyly. It occurs at a very early age.
Mild to moderate dwarfism is seen. Joints of fingers become enlarged.
Mental retardation is frequently found.
Mode of transmission is not known.
and joints, especially of the fingers, become enlarged. Mental retardation is frequently seen. Genetic transmission is unknown.
References
- Unni KK. Cartilaginous lesions of bone. J Orthop Sci. 2001;6:457–72
- D’Angelo L, Massimi L, Narducci A, Di RC. Ollier disease. Childs Nerv Syst. 2009;25:647–53
- Spranger J, Kemperdieck H, Bakowski H, Opitz JM. Two peculiar types of enchondromatosis. Pediatr Radiol. 1978;7:215–9.
- Ikegawa S, Nagano A, Matsushita T, Nakamura K. Metachondromatosis: a report of two cases in a family. Nippon Seikeigeka Gakkai Zasshi. 1992;66:460–6.
- Le Merrer M, Fressinger P, Maroteaux P. Genochondromatosis. J Med Genet. 1991;28:485–9.
- Menger H, Kruse K, Spranger J. Spondyloenchondrodysplasia. J Med Genet. 1989;26:93–9
- Freisinger P, Finidori G, Maroteaux P. Dysspondylochondromatosis. Am J Med Genet. 1993;45:460–4
- . Mellon CD, Carter JE, Owen DB. Ollier’s disease and Maffucci’s syndrome: distinct entities or a continuum. Case report: enchondromatosis complicated by an intracranial glioma. J Neurol. 1988;235:376–8.