Last Updated on September 20, 2023
Myasthenia Gravis is a chronic autoimmune neuromuscular disease that results in varying degrees of weakness of the skeletal muscles of the body. It is a type-II hypersensitivity immune reaction.
The term myasthenia gravis has a Latin and Greek origin. It literally means “grave muscle weakness.”
This weakness increases during periods of activity and improves after periods of rest.
Certain muscles are often, but not always, involved in the disorder. These are
- Eye and eyelid muscles
- Facial muscles
- Chewing muscles
- Muscles involved in talking
- Muscles used in swallowing
- Neck and limb muscles
- Muscles involved in breathing
Drooping eyelids are often the first sign.
Myasthenia gravis affects both genders. It can occur at any age but is more common in those under 40 and over 60 years of age
In less than 40 there is a female preponderance, whereas more males are affected in over 60 group.
The disease is found more commonly than families having other autoimmune disorders.
What Causes Myasthenia Gravis?
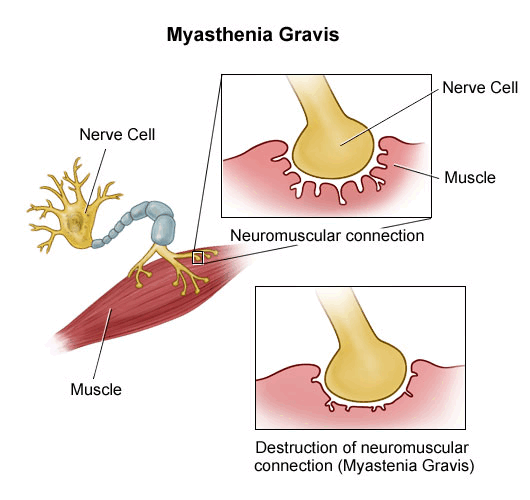
Myasthenia gravis results from antibodies inappropriately attacking and causing injury to certain receptors in muscles that receive nerve impulses. This results in a defect in the transmission of nerve impulses to muscles at the neuromuscular junction where the receptors are located.
Neuromuscular junction, as the name suggests is a specialized structure where nerve cells communicate with muscle cells..
Normally, the nerve endings release acetylcholine, a neurotransmitter that travels through the neuromuscular junction to bind to acetylcholine receptors to cause muscle contraction.
In myasthenia gravis, antibodies or autoantibodies are directed most commonly against the nicotinic acetylcholine receptor (nAChR) in the motor end plate for the neurotransmitter acetylcholine that stimulates muscular contraction.
These antibodies can act in two ways
- Impair the ability of acetylcholine to bind to receptors
- Destroy the receptors.
It has been recently found that the second type of myasthenia gravis is caused actually by antibodies against the muscle-specific kinase, a tyrosine kinase receptor that is required for the formation of the neuromuscular junction.
Thymus gland abnormalities are frequently seen with myasthenia gravis. The thymus gland is involved in immunity function and it naturally grows until puberty. After that, it shrinks. In mysathenia gravis patients thymus gland is found to remain enlarged. Some even have thymomas [tumours of the thymus]. The relation has not been understood completely but the role of the thymus in the formation of autoantibodies is believed to be important.
Associated Conditions
Myasthenia Gravis is associated with various autoimmune diseases, including:
- Thyroid diseases, including Hashimoto’s thyroiditis and Graves’ disease
- Diabetes mellitus type
- Rheumatoid arthritis
- Lupus
- Demyelinating central nervous system diseases
Symptoms and Signs of Myasthenia Gravis
The hallmark of myasthenia gravis is fatiguability. Muscles become progressively weaker during periods of activity and improve after periods of rest. The onset of the disorder can be sudden.
Often symptoms are intermittent.
The diagnosis of myasthenia gravis may be delayed if the symptoms are subtle or variable.
The most commonly affected muscles are the one that controls eye and eyelid movement, facial expression, and swallowing are most frequently affected.
Eyes
Usually, the first noticeable symptom is a weakness of the eye muscles in most of the cases. This may result in drooping of eyelids [ptosis], blurring of vision or double vision [diplopia].
Face Muscles
- Change in facial expressions
- Facial paralysis
Throat
Difficulty in
- Talking [Hoarseness of voice]
- Breathing
- Chewing and swallowing
Back and Chest
- Difficulty in holding neck
- Difficulty in breathing
- Can cause respiratory failure in severe cases due to paralysis of chest muscles and diaphragm
Limbs
- Weakness in upper limb and hand
- Leg weakness causes unstable or waddling gait
All the persons do not have all the symptoms. Even in the same person, the degree of weakness of muscles will vary on days.
The severity of the symptoms varies ranging from a localized form, limited to eye muscles (ocular myasthenia), to a severe or generalized form in which many muscles – including respiratory [That control breathing] are affected.
Myasthenic crisis
It is caused by paralysis of the respiratory muscles and necessitates assisted ventilation to sustain life. It could be triggered by an infection, medication or stress.
Few drugs are known to worsen the symptoms of myasthenia gravis. Some of these are
- Antibiotics – macrolides, fluoroquinolones, aminoglycosides, tetracycline
- Chloroquine
- Beta-blockers
- Calcium channel blockers
- Lidocaine, procainamide
- Trimethaphan
- Diphenylhydantoin
- Lithium
- Chlorpromazine
- Muscle relaxants
- Levothyroxine
- Adrenocorticotropic hormone (ACTH)
- Corticosteroids
A drug history, therefore becomes important.
Classification of Myasthenia Gravis
The most widely accepted classification of myasthenia gravis is the Myasthenia Gravis Foundation of America Clinical Classification
- Class I- Any eye muscle weakness, possible ptosis, no other evidence of muscle weakness elsewhere
- Class II- Eye muscle weakness of any severity, mild weakness of other muscles
- Class IIa: Predominantly limb or axial muscles
- Class IIb: Predominantly bulbar and/or respiratory muscles
- Class III- Eye muscle weakness of any severity, moderate weakness of other muscles
- Class IIIa: Predominantly limb or axial muscles
- Class IIIb: Predominantly bulbar and/or respiratory muscles
- Class IV- Eye muscle weakness of any severity, severe weakness of other muscles
-
- Class IVa: Predominantly limb or axial muscles
- Class IVb: Predominantly bulbar and/or respiratory muscles
- Class V- Intubation needed to maintain airway
Diagnosis of Myasthenia Gravis
Because the symptoms are intermittent, patients are absolutely fine in between two episodes and the symptoms may overlap with other conditions, the diagnosis of myasthenia gravis is usually delayed.
A detailed history can raise the clinician’s suspicion. A detailed examination is done to check for the neuromuscular system including muscle tone, reflexes, and sensory nervous system.
Muscle fatigability when present, strongly suggests the likelihood of myasthenia gravis.
Muscle fatigability
It can be tested by repetitive use of muscles. For example
- For eye muscles
- Looking upward and sidewards for 30 seconds
- Looking at the feet while lying on the back for 60 seconds
- Keeping the arms stretched forward for 60 seconds
- 10 deep knee bends
- Walking 30 steps on both the toes and the heels
- 5 sit-ups
The peek sign indicates the involvement of the ocular orbicularis muscle. After initial apposition of the lid margins, they quickly (within 30 seconds) start to separate and the sclera starts to show.
Lab Studies
Antibodies against the acetylcholine receptor may be found in the serum. A proportion of the patients without antibodies against the acetylcholine receptor may show antibodies against the MuSK protein.
No laboratory tests are available in a time frame that is useful to confirm the emergency diagnosis of myasthenia gravis (MG). An arterial blood gas determination can help guide respiratory management.
Imaging
Chest radiography is indicated to determine the presence of aspiration or other pneumonia, which commonly occurs in patients with myasthenia gravis. CT scan or MRI of the chest is highly accurate in detecting thymoma.
Special Tests
Tensilon (edrophonium) challenge test
- This approach requires the intravenous administration of edrophonium chloride or Tensilon(r)
- This drug inhibits the breakdown of acetylcholine
- Temporarily increases the levels of acetylcholine at the neuromuscular junction.
- The patient shows a dramatic improvement in muscle strength and other features
- The test needs to be done under supervision and monitoring
The test is not completely specific as other conditions like amyotrophic lateral sclerosis may also respond to edrophonium with increased strength.
This test is also used to differentiate between a myasthenic crisis from a cholinergic crisis.
Ice pack test
In a patient with myasthenia gravis who has ptosis, placing ice over an eyelid will lead to a cooling of the lid, which leads to improvement of the ptosis. This occurs because cooling improves neuromuscular transmission.
Electromyography
Electromyography can detect impaired nerve-to-muscle transmission. Muscle fibres in myasthenia gravisdo not respond as well to repeated electrical stimulation compared to muscles from normal individuals.
Spirometry
Lung function testing may be performed to assess respiratory function if there are concerns about a patient’s ability to breathe adequately.
Muscle biopsy
It is only performed if the diagnosis is in doubt and a muscular condition is suspected.
Treatment of Myasthenia Gravis
Emergencies in myasthenia gravis are always treated in hospital settings.
Patients in frank respiratory arrest should be intubated and ventilated.
Any cause for exacerbation should be found and managed. Acetylcholine inhibitors should be given to control muscle weakness.
Treatment of myasthenia gravis on a routine basis is as below
Acetylcholinesterase inhibitors
- Neostigmine and pyridostigmine
- Improve muscle function by slowing the natural enzyme cholinesterase that degrades acetylcholine in the motor end plate
- Thus, the neurotransmitter is therefore around longer to stimulate its receptor.
Immunosuppressive drugs
- Suppress the immunity
- Steroids, cyclosporine, mycophenolate mofetil
Immunomodulators
- Azathioprine
- Mechanism of action against autoimmune disease not clear
Monoclonal Antibodies
- Rituximab
- For adults who are anti-acetylcholine receptor (AchR) antibody-positive
Fc Receptor Antagonists
These lead to increased destruction of IgG which benefits in IgG autoantibody-mediated diseases.
These drugs are indicated in myasthenia gravis patients with ACh receptor antibody
- Efgartigimod – iv infusion over 1 hour
- Efgartigimod/hyaluronidase SC – Subcutaneous infusion
- Rozanolixizumab – Subcutaneous infusion
Plasmapheresis and Intravenous immunoglobulins
If the myasthenia is serious, plasmapheresis can be used to remove the putative antibody from the circulation. Intravenous immunoglobulins can be used to bind the circulating antibodies. Both of these treatments have relatively short-lived benefits
Thymectomy
Thymectomy, the surgical removal of the thymus, is essential in cases of thymoma in view of the potential neoplastic effects of the tumor. Most of these patients show significant improvement after the thymus is removed.
The role of removal of the thymus is not very clear in patients who do not have an abnormality of the thymus.
Prognosis
With treatment, the outlook for most patients with myasthenia gravis is bright with a normal life expectancy.
Quality of life can vary depending on the severity and the cause.
Most patients need treatment for life.
The disease does not worsen in severity with age.
Complications
- Respiratory failure
- Most severe complication
- Presents as the rapid deterioration of respiratory effort
- Ultimately apnea occurs
- Pneumonia
- Chronic respiratory insufficiency
- Cholinergic crisis – Due to an overdose of acetylcholinesterase overdose or other drugs.
- Steroids adverse effects
- Opportunistic infections, renal insufficiency, and hypertension due to immunosuppressive medications
Myasthenia Gravis In Children
Myasthenia gravis most commonly affects adults and the elderly, but it has been known to occur at any age. Presentation and causes of the myasthenia-like picture in children differ from adults.
Three types of myasthenic symptoms in children can be distinguished.
- Neonatal Myasthenia Gravis
- It occurs in 12% of pregnancies with a mother with myasthenia gravis.
- In such cases, the mother passes the antibodies to the infant through the placenta causing neonatal myasthenia gravis.
- The symptoms will start in the first two days and disappear within a few weeks after birth.
- Congenital Myasthenic Syndrome
- Otherwise healthy mother
- Develop myasthenic symptoms beginning at birth.
- Due to synaptic malformation, which in turn is caused by genetic mutations.
- Hereditary disease and different from myasthenia gravis
- Typically autosomal recessive.
- Juvenile Myasthenia Gravis
- It is myasthenia that occurs in childhood.
References
- Jayam Trouth A, Dabi A, Solieman N, Kurukumbi M, Kalyanam J. Myasthenia gravis: a review. Autoimmune Dis. 2012;2012:874680. doi: 10.1155/2012/874680.
- Gold R, Schneider-Gold C. Current and future standards in treatment of myasthenia gravis. Neurotherapeutics. 2008 Oct. 5(4):535-41