Last Updated on October 30, 2023
Paget disease of bone was first described by James in 1877 who described it as chronic inflammation of the bone and called it osteitis deformans.
The term osteitis deformans is now considered technically incorrect, and the preferred term is osteodystrophia deformans.
Paget disease is a localized disorder of bone remodeling that typically begins with excessive bone resorption by osteoclasts, followed by an increase in bone formation by osteoblasts.
This leads to a structurally disorganized bone which is weaker, larger, less compact, more vascular, and more susceptible to fracture than normal adult lamellar bone.
There is a juvenile Paget disease also which is characterized by widespread skeletal involvement and has different histologic and radiologic features.
The prevalence of Paget disease ranges from 1.5 to 8.0 percent.
Europe has got the highest prevalence of this disease.
It is almost twice as common in males as females.
Paget disease increases in frequency with increasing age. It is is most commonly diagnosed in the sixth decade.
Monostotic [affecting single bone] disease accounts for 10-35% of cases.
Pathophysiology of Paget Disease
Three phases of the disease have been identified- lytic, mixed lytic and blastic, and sclerotic.
Different bones in the same individual may be in different phases of the disease.
Lytic phase
In this phase osteoclasts [cells that resorb the bone] increase in number and resorb the normal bone. These osteoclasts are larger in size and have many more nuclei than normal osteoclasts.
Mixed Phase
In the mixed phase, osteoblasts [bone forming cells] form the bone in reaction to resorption of the bone. The newly made bone is abnormal woven bone and haphazardly laid. Due to increased activities of osteoblasts and osteoclasts, bone turnover increases.
Sclerotic phase
With time bone resorption by osteoclasts decreases but abnormal bone formation continues. Woven bone, in some pockets, may be replaced by normal lamellar bone.
Eventually, bone formation declines and the condition become quiescent. This is the sclerotic, or burned out phase. There is a minimal activity of the bone in this phase.
Bones Affected by Paget’s Disease
Paget disease can affect every bone in the skeleton, but it has an affinity for the axial skeleton, long bones, and the skull. The skeletal sites primarily affected include the pelvis, lumbar spine, femur, thoracic spine, sacrum, skull, tibia, and humerus. The hands and feet are very rarely involved.
Etiology [Cause] of Paget Disease
The etiology of Paget disease is unknown, both genetic and environmental contributors have been suggested. However exact cause is unkown yet.
Genetic Factors
A positive family history has been reported in 12-22% people with Paget’s disease with 7- to 10-fold increase in the incidence in relatives of patients diagnosed with the disease.
Few studies also have suggested an association between human leukocyte antigen [antigen on white blood cells]. Other studies have found clinical evidence of disease and mutations in the sequestosome SQSTM1/p62 in 30% of familial Paget cases.
Cytokines, RANKL [Receptor activator of nuclear factor kappa-B ligand] induced bone resorption and macrophage colony stimulating factor have been suggested to play a role however exact role remains to be determined.
Environmental Factors
Because there is a variation of expression of disease within the families, environmental factors may be responsible.
Viral Infection
According to this hypothesis, bone marrow cells are infected by a virus and lead to an abnormal increase in osteoclast formation. This virus may take years, which may account for the advanced age of most people diagnosed with Paget disease. However, till date, the theory has not been proven.
Some studies have found viral inclusion particles in pagetic osteoclasts. However, the presence of these inclusions may be markers of the disease itself rather than causative agents.
Other Etiologies
The possibility of an inflammatory cause of Paget disease is supported by evidence of clinical improvement after treatment with anti-inflammatory medications. Autoimmune, connective tissue and vascular disorders are proposed as other possible etiologies.
Presentation
Most persons with Paget disease do not have any symptom. An incidental deranged lab value like an elevated serum alkaline phosphatase level or findings on x-ray may lead to a diagnosis.
Dull, deep, boring pain is the most common presenting symptom. The pain may exacerbate during the night. The site of the pain depends on the bone involved. For example, the patient may present with hip pain in case acetabulum or upper femur is involved.
Pain on weight bearing may result due to bony deformities or effects of bone softening like protrusio acetabuli.
Altered stresses may lead to pain in the joints.
Patients may also present with of complications also. These include musculoskeletal, neurologic, and cardiovascular complications and are discussed separately later in the article.
Pathologic fractures, nonspecific headaches, impaired hearing, and tinnitus are common presentations due to complications of the diseases. Increase in head size, skull deformity may occur in severe disease.
The most common cranial symptom is hearing loss, occurring in 30-50% of patients with skull involvement. Changes in vision may occur secondary to optic nerve involvement.
Back and neck pain are common complaints due to the involvement of the spine. Facial disfigurement and malocclusion may occur in jaw bone involvement. Ataxia, gait disturbances, dementia, and neurological compromise may result from hydrocephalus and cerebellar compression.
The most dreaded complication is neoplasm and it should be suspected in increased bone pain with an enlarging soft tissue mass.
Physical examination may reveal the patient to be normal. Bony deformities, such as an enlarged skull, spinal kyphosis, and bowing of the long bones of the extremities may be present. Patients with Paget disease may also have gouty arthritis and should be evaluated for.
Localized pain, warmth and tenderness may be elicited with manual palpation. Bruits of the tibia or skull may be noted in severe cases.
A tender soft tissue mass may be seen in a malignant tumor.
Complications of Paget disease
Fractures
Incomplete stress fractures may occur in Paget disease. Mild injuries may cause acute true pathologic fractures in the weakened pagetic bone. Pathologic fractures are more common in women than in men.
The site of these fractures is the femur, tibia, humerus, spine, and pelvis. Subtrochanteric region, followed by the upper third of the femoral shaft and neck of the femur are common areas of fractures.
Another problem with fractures in Paget disease is nonunion [up to 40%] and refracture at the same sites are much more common due to the disease affecting developing callus.
Malignancy
It should be suspected in patients who have a sudden increase in bone pain or swelling. Sarcoma of Paget disease has a rapid, fatal course. Malignant change occurs more in patients with an extensive disease than not extensive.
Men have a slightly higher incidence of sarcomatous degeneration than women. The femur is the most commonly affected site, followed by the proximal humerus.
The peak incidence of sarcoma in Paget’s disease is in 7th and 8th decade.
Osteosarcoma is the most common type of pagetic sarcoma, followed by fibrosarcoma, chondrosarcoma, and sarcoma of myeloid and mesenchymal elements.
Malignant change in Paget disease should be suspected in the following situations.
- Increased pain with a progressive lytic lesion
- An enlarging soft tissue mass
- Bony speculation
- Persistent fracture without healing
- Cortical destruction
Pathological fracture of the bone is the presentation in one-third of sarcoma cases. In 33% of the cases, the presentation involves a pathological fracture of an affected long bone. Biopsies of pathological fractures may be recommended to rule out sarcoma.
Giant cell tumors are benign tumors that may occur in Paget’s disease. They usually involve the facial bones and mandible present as a soft tissue mass with a lytic lesion.
Neural Complications
Apart from injury due to a fractured vertebra, spinal cord injury may occur due to enlargement of the pedicle, lamina, and other structures. Nerve root or spinal nerve compression may also occur. Spinal cord compression is most frequent in the upper thoracic spine.
Platybasia may lead to spastic quadriplegia. Basilar invagination or compression of posterior fossa structures of the skull may lead to cerebellar or brainstem syndromes.
In the lumbar spine, cauda equina syndrome and spinal canal stenosis may occur. Hydrocephalus is rare.
Entrapment of cranial nerves by pagetic bone may result in the expected cranial nerve palsies, the most common being vestibulocochlear nerve. The optic nerve may be the second most commonly affected cranial nerve.
Joint Disease
Hip, knee and glenohumeral joint are mostly affected joints where degeneration occurs in Paget disease. In degenerative joint disease in Paget disease, osteophyte formation is not prominent. The pathophysiology of arthritic changes associated with Paget disease is unknown.
Other complications of Paget disease include the following:
- Hashimoto thyroiditis
- Dupuytren contracture
- Chondrocalcinosis
- Osteogenesis imperfecta
- Osteopetrosis
- Kidney stones
- Loosening of teeth
- Angioid streaks
Associated Conditions
There are conditions that have been found to coexist with Paget disease but are not due to Paget disease per se
- Rheumatoid arthritis
- Psoriatic arthritis
- Ankylosing spondylitis
- Diffuse idiopathic skeletal hyperostosis
- Pseudogout
- Peyronie disease
- Pigmented villonodular synovitis
- Hyperuricemia and gout
Cardiovascular Abnormalities
Increased cardiac output, left ventricular hypertrophy to occur due to an increase in the blood supply of the bone. High-output congestive heart failure may occur in severe widespread disease. Calcific aortic stenosis and interventricular septum may also occur.
Lab Studies
Standard values for serum calcium, phosphorus, and parathyroid hormone levels, are usually normal in Paget disease.
Raised levels of uric acid have been reported in patients with Paget disease.
Lab diagnosis of Paget disease is based on assay of markers of bone resorption and formation. These markers also help in monitoring the treatment response.
Upon successful treatment of Paget disease, the level of these bone markers is expected to decrease.
Alkaline Phosphatase
Both total and bone-specific alkaline phosphatase levels are increased due to bone formation. Alkaline phosphatase reflects the disease activity. However, normal alkaline phosphatase level does not exclude the disorder because in monostotic disease or local disease, the total alkaline phosphatase level may be normal. In such cases, bone-specific alkaline phosphatase should be ordered.
Bone-specific alkaline phosphatase is good for assessing Paget disease activity and has the highest sensitivity for diagnosis [84%] followed by alkaline phosphatase [74%] and is considered as a marker of bone formation.
Serum calcium and phosphate levels should be within the reference range in patients with Paget disease. Urinary excretion of calcium also should be normal.
Raised levels of calcium in blood and urine may occur in case of immobilized patients or coincident primary hyperparathyroidism.
Urinary Markers
- Pyridinium collagen cross-links, pyridinoline and deoxypyridinoline(a marker of bone resorption)
- N -Telopeptide of type I collagen (a marker of bone resorption)
- Alpha-alpha type I C -telopeptide fragments (a marker of bone resorption)
Urinary hydroxyproline levels reflect increased bone resorption due to osteoclastic activity and are increased in Paget’s disease. Hydroxyproline is a product of collagen breakdown 20-30% of total hydroxyproline levels are from bone resorption. Hydroxyproline levels have been correlated with the extent and activity of the disease. Because diet may affect the levels, overnight fasting is necessary before testing.
This investigation is not reliable in patients with skin disease.
Urine levels of excreted bone-specific pyridinium collagen cross-links, pyridinoline and deoxypyridinoline, is considered sensitive, specific and better indicators of bone resorption and response to treatment than the hydroxyproline assay.
Procollagen I N -terminal peptide (PINP) has emerged as a sensitive serum marker for bone formation.
Serum osteocalcin, which is produced specifically by osteoblasts, does not reflect disease activity.
Uric Acid
Hyperuricemia in Paget disease is caused by the increased turnover of nucleic acids from high bone turnover•
Secondary hyperparathyroidism may occur in 10-15% of patients with Paget disease. Inadequate calcium intake in the face of increased demand from extensive bone remodeling may lead to increases parathyroid hormone.
Imaging
X-rays
X-rays should be done at the time of initial evaluation. The skeletal survey can be done to look for multiple bony involvements but bone scan scintigraphy is more sensitive, safer and better alternative.
Repeated radiography may be needed to monitor arthritic changes or evaluating for malignant degeneration or fracture.
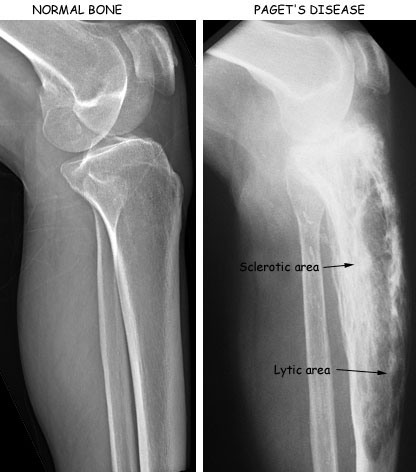
In early disease, radiographs may show lytic lesions due to bone resorption and in later areas of increased density [sclerosis] or as coarsened trabecula due to repair attempt.
Lysis may be absent in later disease and only sclerosis may be seen. Bones may get enlarged and widened.
Xrays of long bone show osteolysis of the subchondral region [ just near the joint], with extension into the metaphysis and diaphysis.
Advancing osteolysis may appear as a V- or wedge-shaped radiolucent area. Focal radiodensities with cotton wool appearance can be noted.. Areas of lysis and radiodensities may be separate or superimposed.
In the pelvis, iliopectineal line thickening may occur [Brim sign]
Paget disease of the spine affects the vertebral bodies and posterior elements. Radiographs reveal enlarged coarse trabeculae combined with the prominent radiodense peripheral contour of the vertebral body [ Picture frame appearance]
A homogeneous increase in osseous density in the vertebral body can be seen [ Ivory vertebra]. The ivory vertebra is also seen skeletal metastasis and lymphoma
Weak pagetic bone results in biconcave-shaped vertebral bodies [fish vertebra] due to disc compression of the weakened vertebrae. These are also seen in other bone softening diseases like [osteomalacia, hyperparathyroidism, and osteoporosis.]
Spinal radiographs may also show a decrease in intervertebral disc space due to degenerative disc and joint disease. Loss of vertebral heights and fusion of vertebrae are other findings.
MRI/CT
Computed tomography and magnetic resonance imaging are not needed for the diagnosis of Paget disease of bone. But they are useful in the evaluation of complications like malignancy, joint abnormalities, and spinal problems.
CT scanning and MRI are useful to diagnose and evaluate neurologic complications, such as basilar invagination, spinal cord compression, or hydrocephalus. Spinal stenosis and vertebral involvement are assessed best with CT scanning or MRI.
CT scanning provides better visualization of bone and MRI gives superior detailing of the brain, spinal cord, cauda equina, and soft tissue.
Malignant changes and their extent of are evaluated better with MRI.
Bone Scans
These are helpful in Paget disease to find the extent of the disease and in succession effects of the treatment.
Bone scanning is sensitive but not as specific as x-ray. In the quiescent osteosclerotic stage, lesions may be detected radiographically but not with bone scans.
Histology
Not required routinely. It is indicated for sarcomatous changes or nonhealing fracture to rule out malignancy in Paget disease.
Treatment of Paget Disease
The immediate goal of the treatment is to control the disease activity. Long-term goals include to slow/prevent the progression of the disease, and decrease complications.
Drug therapy for Paget disease is
-
- Bisphosphonates
- Calcitonin
- Analgesics
Calcium and vitamin D supplements
Bisphosphonates
Bisphosphonates are analogs of inorganic pyrophosphate and act by binding to hydroxyapatite in bone matrix, thereby inhibiting the dissolution of crystals.
They prevent osteoclast attachment to the bone matrix and osteoclast recruitment and viability.
Following bisphosphonates are used for the treatment of Paget Disease along with serial monitoring of bone markers to check the response. Bisphosphonates are first-line drugs for Paget disease. Bisphosphonates suppress the resorption of the bone.
- Pamidronate
- Alendronate
- Tiludronate
- Risedronate
- Zoledronic acid
Response to therapy is indicated by the reduction of symptoms and decreases in levels of bone-specific alkaline phosphatase (a bone formation marker) and deoxypyridinoline, C -telopeptide, or N -telopeptide (bone resorption markers).
Etidronate has been found to be less effective than the other bisphosphonates in suppressing biochemical markers of disease activity and should not be used as a first-line agent if the other bisphosphonates are available
If a patient has not responded after 6 months following treatment or if clinical or biochemical relapse occurs, retreatment with a change of drug is indicated.
If the pain is not relieved, persistence or relapse is a possibility but disease activity should be confirmed. If not, other sources of pain should be investigated.
Orthotic devices, including canes, braces, and walkers, may be useful for patients with gait abnormalities resulting from Paget disease that involves the lower limbs.
Chemotherapy, radiation, or both may be used to treat neoplasms that arise from the pagetic bone. Amputation may also be necessary.
Calcitonin
This agent is a peptide hormone that binds to calcitonin receptors on osteoclasts and rapidly inhibits bone resorption. It also has a significant pain-relieving effect on bone. Human calcitonin is no longer available and available salmon calcitonin is used. It is a synthetic copy of a polypeptide hormone secreted by the ultimobranchial gland of salmon. Patients treated with salmon calcitonin demonstrate decreased responsiveness after initial improvement due to antibody formation against the drug.
Calcitonin is also associated with cancers.
Symptoms including bone pains and warmth improve and markers respond favorably with calcitonin. Restoration of more normal bone can be seen radiographically, especially after chronic calcitonin treatment.
Calcitonin is administered by injection, three times per week or daily, for 6–18 months. Repeat courses can be given after brief rest periods.
However, if no response is noted by 3 months, treatment should be discontinued.
Calcitonin is seldom used only if bisphosphonates are contraindicated. Calcitonin is also linked to increased chance of cancer and few regulations forbid use beyond three months.
Analgesics
Both nonsteroidal and opioid drugs can be given to relieve arthritic joint pains. Bone pains of Paget disease do not respond to these drugs.
Supplements
Patients should receive 1000-1500 mg of calcium and at least 400 U of vitamin D daily. This recommendation is especially important in conjunction with bisphosphonate treatments.
Surgical Treatment
Neoplasm
Because the tumors are very agggressive, amputation usually is the most appropriate treatment. Amputation has been the most effective surgical management, especially with a spontaneous pathologic fracture. Pagetic sarcomas are associated with poor prognsosis.
Joint Disease
Unstable fractures and severe arthritis refractory to medical and physical therapy would require surgery.
Joint replacement surgery may be indicated for advanced joint disease if nonsurgical treatment fails to relieve pain adequately.
Total hip replacement is the most common orthopedic surgery performed on patients with Paget disease.
Total hip arthroplasties have been quite successful, relieves pain and improves function.
Treatment with bisphosphonates or calcitonin reduces intraoperative bleeding by decreasing disease activity.
Medication should be started at least 6 weeks prior to elective surgery.
Tibial and fibular osteotomies to correct the alignment of the bones are also done for relieving pains of knee and ankle.
After surgery, the patient should be educated about delayed bone healing and a long rehabilitation. Weight bearing should be delayed and protected because of poor strength of Pagetic bone.
Spinal Disease
The most common cause of neurologic dysfunction from the pagetic spinal stenosis is osseous compression from an enlarged vertebral body. Symptomatic pagetic spinal stenosis can be treated successfully with bisphosphonates and calcitonin.
Surgical decompression rarely is needed for spinal stenosis and persistent mechanical pain unresponsive to nonsurgical treatment.
Follow up
Because of the increased risk of malignancy, patients with Paget disease should be monitored indefinitely.
A patient who has a family history of Paget disease and is older than 40 years may have an alkaline phosphatase blood test every 2-3 years. If the alkaline phosphatase level is within the reference range, radiography or bone scanning also may be performed.
Untreated patients with mild disease should be scheduled for annual serum alkaline phosphatase levels and annual radiographs of osteolytic lesions.
Treated patients should have serum alkaline phosphatase levels every 3-4 months and should undergo annual radiographs of osteolytic lesions, if present. Alternatively, urinary hydroxyproline or collagen cross-links can be used.
Prognosis
The general outlook for patients with Paget disease is good, especially if treatment is administered before major changes have occurred in the bones. Treatment does not cure Paget disease, but it can control it. Patients with multiple bony involvements have a less favorable prognosis.
The prognosis is extremely unfavorable with sarcomatous change and the 5-year survival rate is 5-7% only.
Patient Education in Paget Disease
Patients should be educated about proper posture, body mechanics, and avoidance of trauma. The patient should stay active but at the same time take measures to avoid falls.
Patients should be aware of the signs and symptoms of complications is important. For example, increased local pain with soft tissue mass may represent a sarcoma.
Patient education about delayed bone healing and the long rehabilitation process is important in situations of fracture and postsurgery. Reinforcement about the importance of careful, prolonged, protected weight bearing is crucial because the pagetic bone is abnormal and weak.
Family members should be told about likelihoood of disease in relatives and as mentioned above can be tested for markers of the disease.
Juvenile Paget disease
Juvenile Paget disease is a very rare disease which is known by many other names as well. These are
- Chronic congenital idiopathic hyperphosphatasemia
- Familial idiopathic hyperphosphatasemia
- Familial osteoectasia
- Hyperostosis corticalis deformans juvenilis
- Hyperphosphatasemia with bone disease
- Familial idiopathic hyperphosphatasia
- Idiopathic hyperphosphatasia
- Osteochalasia desmalis familiaris
- Osteoectasia with hyperphosphatasia
Juvenile Paget disease is a condition where there is an increase in bone turnover leading to bone pain bony deformities. This disease causes bones to enlarge, deform and easily broken. The juvenile form of Paget’s disease is different from the adult Paget disease.
This condition is inherited in an autosomal recessive manner, which means both copies of the gene from each parent has to contribute. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene without showing signs and symptoms.
Known gene locus is 8q24. The disease results from mutations or deletions in the TNFRSF11B gene. This mutation leads to osteoprotegerin which is an inhibitor of osteoclast differentiation and bone resorption.
This leads to increased osteoclast differentiation which in turn lead to increased bone resorption. Bone tissue is resorbed more quickly and in response to the bone tissue that grows it is larger, weaker, and less organized than normal bone.
Juvenile Paget disease is a very rare condition and only about 50 people are affected till now worldwide.
Presentation
The patient with Juvenile Paget disease present in early childhood
- Bone pain and bone deformities [Bowed limbs]
- Failure to thrive.
- Dwarfism.
- Progressive enlargement of the head.
- Difficulty walking.
- Pigeon chest deformity.
- Muscular weakness.
- Premature shedding of the teeth.
These abnormalities become more severe during the adolescent growth spurt when bones grow very quickly.
Thickening of the skull bones can lead to hearing loss. Deformation and collapse lead to abnormal curvature of the spine.
Weight-bearing long bones in the legs tend to bow and fracture easily.
Retinal degeneration with or without angioid streaks, renal stones, and internal carotid aneurysm are known associated findings
Alkaline phosphatase is raised. Urinary excretion of hydroxyproline is raised.
Enlargement and thickening of bones of skull vault are noted on x-rays.
A healthy diet should be maintained for children. Adequate physical activity should be maintained.
Bisphosphonates and calcitonin are associated with clinical, biological and radiological improvement.
Recombinant versions of osteoprotegerin are under research for managing this condition.
Following complications are know with Juvenile Paget Disease
- Bone pain
- Pathological fractures
- Kyphoscoliosis of spine
- Acetabular protrusion – protrusion of acetabulum into the pelvis.
- Deafness
- Osteoporosis
- Delayed motor development
The severity of the disease increases as the child progresses towards adolescence. Some patients will survive beyond the age of 50 years.
Sources For Patient
- The Paget Foundation